Turner syndrome is a chromosomal disorder that affects females and is caused
by a complete or partial loss of the second sex-determining chromosome. The syndrome
is typically characterized by short stature, ovarian insufficiency, and
malformations in organ systems that could include cardiac defects (particularly
coarctation of the aorta and bicuspid aortic valve), lymphedema (especially nuchal
and over the dorsum of the hands and feet), short 4th metacarpals, and genitourinary
malformations (such as horseshoe kidney). Some affected individuals are
phenotypically normal females with only short stature. Others can have
life-threatening cardiovascular, hormonal, and lymphatic anomalies or
manifestations, such as short stature, pubertal delay, and sterility, which impart
significant psycho-emotional burden and a higher risk for co-morbidities.
Missing link with id: asthma
Other Names & Coding
Bonnevie-Ullrich-Turner syndrome
Gonadal dysgenesis
Ovarian dysgenesis
Turner's syndrome
Ullrich-Turner syndrome
XO syndrome, monosomy X
Turner syndrome is present in approximately 1 in 2000 to 2500 live female births
worldwide. [Donaldson: 2006]
[Stochholm: 2006] Prevalence is greater if pregnancies that
do not survive to term are taken into account; one study showed that 66% of
pregnancies affected by Turner syndrome resulted in spontaneous miscarriage.
[Iyer: 2012]
Genetics
About half of women with Turner syndrome are completely missing the second sex
chromosome; in the other half, it may be partially missing or rearranged. Mosaic
Turner syndrome occurs when only some of the individual’s cells lack the second
normal sex chromosome. The severity of the phenotype is related to the absence or
presence of a second sex chromosome. Full monosomy (45,X or 45,XO) typically is the
most severe form and mosaic Turner syndrome is typically the mildest
form.
Prognosis
Congenital and atherosclerotic heart disease increases risk of mortality. Health
complications include type 2 diabetes and osteoporosis. Girls and women with Turner
syndrome have a small risk for fatal aortic dissection, which is greater in the
presence of abnormalities of the aorta or aortic valve and hypertension. Girls with
Turner syndrome may have normal intelligence, but they are at risk for social
immaturity, attention-deficit disorder, and learning disabilities. [Ostberg: 2003]
Practice Guidelines
Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, Lin AE, Mauras N, Quigley CA, Rubin K, Sandberg DE,
Sas TCJ, Silberbach M, Söderström-Anttila V, Stochholm K, van Alfen-van derVelden JA, Woelfle J, Backeljauw PF. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International
Turner Syndrome Meeting. Eur J Endocrinol.
2017;177(3):G1-G70.
PubMed abstract
Roles of the Medical Home
A child with Turner syndrome needs a medical home for well-child and
chronic-care visits and to facilitate and coordinate access to subspecialists (most
often including cardiologists and endocrinologists) for monitoring and early
intervention when applicable. The medical home may also help to reduce duplication
of services and unnecessary medical appointments (which place unnecessary emotional
and financial burden upon families).
Clinical Assessment
Overview
Turner syndrome may be diagnosed
across the lifespan. The broad clinical spectrum of TS ranges from a classic
appearance with many different physical features to no apparent or minimal
observable features. Growth failure is a problem for virtually all. Turner syndrome
patients need ongoing health surveillance for co-morbidities throughout their life.
Ideally, a multidisciplinary clinic or team of specialists with expertise in Turner
syndrome would coordinate and manage care.
Pearls & Alerts for Assessment
High false positive prenatal results
As many as 30% of fetuses suspected of having Turner syndrome based on
cytogenetic studies alone may ultimately have normal karyotypes and normal
physical exam findings. Prenatal diagnosis of Turner syndrome should always
be accompanied by fetal ultrasonography; the likelihood of Turner syndrome
is reduced if sonographic results are normal (e.g., no cystic hygroma,
renal, or cardiac malformations). [Gravholt: 1996]
[Huang: 2002]
Risk of gonadoblastoma
Any patient with Turner syndrome who has marker chromosome elements (sex
chromosome material of uncertain origin) detected on the karyotype or who
develops virilization should be screened for Y chromosome mosaicism. The
presence of a Y chromosome can be detected by standard karyotype or PCR
(polymerase chain reaction). When Y-chromosome material is present
(incidence of 5–12%), prophylactic gonadectomy is recommended due to an
increased risk (~10%) of gonadoblastoma. [Binder: 1995]
[Gravholt: 2017]
Short stature and Turner syndrome
While individuals with Turner syndrome may exhibit a constellation of unusual
features and organ malformations, short stature and growth failure are the
only findings that are present in virtually 100% of patients. [Palmer: 1976]
Aortic dissection and rupture risk
The incidence of acute aortic dissection in young and middle-aged women with
Turner syndrome is more than 100 times that of the general population, and
the mean age of onset in those with Turner syndrome is 30 years (range 4-64)
compared to 71 years. [Bondy: 2008]
[Carlson: 2007] Risk factors for dissection include
systemic hypertension, aortic dilatation, and aortic valve abnormalities,
though only rarely is a predisposing factor identified. Presenting
complaints may be vague or minor in nature, such as abdominal pain,
"heartburn," back or shoulder pain, or a change in voice (related to
traction on the recurrent laryngeal nerve). If persistent, these symptoms
should be given serious investigation, including trans-esophageal
echocardiography,chest CT, or cardiac MRI.
Screening
For the Condition
Although screening for Turner syndrome in newborn infants is not
recommended, prenatal cytogenetic screening for aneuploidy is increasingly
performed for mothers of advanced maternal age, and cytogenetic evidence of
Turner syndrome can be an incidental finding. Ultrasonography can play an
important role in diagnosing Turner syndrome in utero. Increased nuchal
translucency is common in Turner syndrome fetuses, but it is also seen in the
autosomal trisomy syndromes. The presence of a frank cystic hygroma, however,
makes Turner syndrome diagnosis more likely. Other
ultrasound findings such as coarctation of aorta or cardiac defects,
intrauterine growth restriction, renal anomalies, brachycephaly, poly or
oligohydramnios are further suggestive of Turner syndrome. If the results of
fetal ultrasonography are normal, fetal cytogenetic studies have a relatively
high false positive rate and non-invasive prenatal testing (NIPT) yields 2.5 as
many false positives for Turner syndrome as it does true positives. [Yu: 2017] These results should be considered with caution when
counseling a family about the risk of Turner syndrome. Regardless of the test
procedure or results, genetic counseling should be provided before and after any
prenatal diagnostic procedure. Ultrasound and maternal serum screening are not
diagnostic, and karyotype confirmation of Turner syndrome is absolutely needed.
Advanced maternal age is not a risk factor for Turner
syndrome.
Of Family Members
The risk of the same parents having a second child with Turner syndrome is
no greater than that of the general population - no screening is
recommended.
For Complications
Turner Syndrome Health Maintenance Checklist ( 80 KB)Click image
for full-size pdf.
Numerous specific screens are recommended; they are summarized below
and detailed in a checklist format for use in practice (left).
Annual physical exam, including height, weight, blood
pressure, and skin exam
Comprehensive ophthalmologic exam starting at 12-18
months or at diagnosis
Audiometric evaluation every 3 years starting at 9-12
months of age
Scoliosis/orthopedic evaluation annually
Renal ultrasound at diagnosis
Cardiovascular:
Resting EKG and QTc measurement at diagnosis
Transthoracic echocardiogram (TTE) at diagnosis
Cardiac MRI (CMR) as soon as feasible without
need for general anesthesia
TTE or CMR (click on charts below for details)
In the absence of aortic abnormalities,
every 5 years
If hypertension (HTN), bicuspid aortic valve (BAV), coarctation of
the aorta (CoV), or Turner syndrome-specific aortic size
Z-score (TSZ) >3, yearly
After 16 years, frequency determined by risk
Cardiac screening in Turner syndrome, infant - 16
yrs. (from [Gravholt: 2017]) Click image for
full-size version.
Cardiac screening in Turner syndrome, above 16 yrs. (from
[Gravholt: 2017]) Click image for full-size
version.
Thyroid function tests annually starting at age 4
Liver function tests annually starting at age 10
Annual HbA1c, glucose starting at age
10
Neuropsych evaluation at preschool and school
transitions (to high school and higher education)
Pediatric dental specialist by age 2, orthodontic
evaluation no later than age 7
Dermatology follow up for nevi
Nutritional evaluation, including celiac screening
every 2 years, starting at age 2
25-OH vit D level between 9-11 years and every 2-3
years thereafter
Presentations
Epicanthal FoldsMissed and delayed diagnosis of Turner syndrome remain a problem.
[Gravholt: 2017] Regardless of other findings, Turner
syndrome should be considered in any female with unexplained growth failure or
delayed puberty. Presentations of Turner syndrome may include:
Unexplained growth failure
Low-set ears
Micrognathia
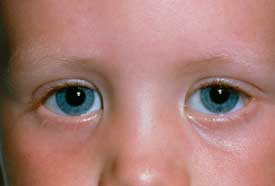
Epicanthal folds
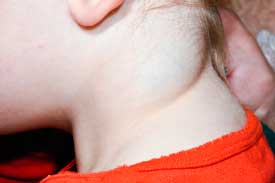
Nuchal redundancy, cystic hygroma
Widely spaced nipples, perhaps with shield chest and pectus
excavatum
Cystic HygromaLymphedema sequence (edema of hands or feet, webbed neck, low
hairline, hyperconvex and hypoplastic nails)
Cardiac anomalies, such as bicuspid aortic valve,
coarctation of aorta
The diagnosis of Turner syndrome requires the presence of characteristic
features in phenotypic females and full or partial sex chromosome monosomy, with or
without cell line mosaicism, demonstrated on a standard 20-cell karyotype.
Additional metaphases may be counted or FISH studies
performed if there is suspicion of undetected mosaicism. Although a peripheral blood
karyotype is usually adequate, a second tissue, such as skin fibroblasts, buccal
mucosa cells, or possibly bladder epithelial cells from the urine, may be examined
if there is a strong suspicion of Turner syndrome and the karyotype is normal. Any
patient with Turner syndrome who has marker chromosome elements (sex chromosome
material of uncertain origin) detected on the karyotype or who develops virilization
should be screened for Y chromosome mosaicism. The presence of a Y chromosome can be
detected by standard karyotype or FISH (fluorescent in situ hybridization) or PCR
(polymerase chain reaction). PCR techniques are more sensitive than FISH in
detecting cryptic Y-material. When Y-chromosome material is present (incidence of
5–12%), prophylactic gonadectomy is recommended due to an increased risk (around
10%) of gonadoblastoma. [Binder: 1995]
[Gravholt: 2017]
45, X (monosomy X) is found in approximately 45% of live
births with Turner syndrome; these patients should be evaluated for presence
of Y chromosome material.
45, X mosaicism is a mosaic chromosomal complement (e.g.,
45,X/46,XX) detectable in 20-30% of all patients with Turner syndrome.
X chromosome anomalies:
Xp or Xq deletion: Some patients have a deletion of
the short arm of the X chromosome with or without 45,X cell line
mosaicism. Patients with terminal Xq deletion, may not have any
other features of Turner syndrome besides ovarian insufficiency.
Ring chromosome X
Isochromosome Xq (46,X,i(X)q): Patients with a
structurally abnormal X chromosome consisting of 2 copies of the
long arm with some intervening short arm or centromeric material are
at higher risk for autoimmune disorders.
Patients with mosaic 46,XX karyotype or isochromosome Xq have a
milder phenotype, while patients with mosaicism for 46,XY cell line or structural
rearrangement of the Y chromosome mostly have masculinized external genitalia and
are at increased risk for having gonadoblastoma and other gonadal tumors.
Differential Diagnosis
Noonan syndrome (NS) is an autosomal dominant condition affecting
boys and girls in equal proportions. It is most commonly associated with pathogenic
variants in the PTPN11, SOS1,
RAF1 or RIT1 genes (Noonan
syndrome next-generation sequencing (NGS) panel testing is clinically available). As
in Turner syndrome, girls with NS typically have short stature, lymphedema of the
extremities, and can have neck webbing, cardiac defects, ptosis, inner epicanthal
folds, high-arched palate, and musculoskeletal differences (pectus deformities,
cubitus valgus). However, the cardiac anomalies most commonly seen in NS are right
sided (pulmonary valve stenosis in 50% of those with cardiac anomalies; septal
defects and cardiomyopathy may also occur); the cardiac anomalies most commonly seen
in Turner syndrome are usually left sided. Individuals with NS are more likely to
have gross motor or global developmental delays than those with Turner syndrome.
Despite these differences, considerable variability of presentations exists for both
syndromes and a karyotype with complete or partial absence of the second
sex-determining chromosome is often the only way to distinguish between the 2
disorders in females. [Cassidy: 2005]
Comorbid & Secondary Conditions
Co-morbidities in Turner syndrome are often undiagnosed and include:
[Freriks: 2011]
Aorto-cardiac anomalies
Dyslipidemia
Renal anomalies
Hypertension
Osteopenia
Hearing loss
Primary ovarian insufficiency
Infertility
Celiac disease
Hypothyroidism
Hepatic fibrosis
History & Examination
Turner syndrome should be considered in any girl with a webbed neck,
lymphedema, or coarctation of aorta during infancy. Previous guidelines
suggested that a peripheral blood karyotype be considered in girls with
unexplained short stature, delayed puberty, or the constellation of
characteristic dysmorphic features [Bondy: 2007], but
new guidelines propose that karyotyping be performed: [Gravholt: 2017]
[Shankar: 2018]
In the presence of a single clinical
feature such as fetal hydrops or cystic hygroma,
unexplained short stature, obstructive left-sided cardiac abnormality
(such as a bicuspid aortic valve, coarctation, aortic stenosis,
hypoplastic left heart syndrome, or mitral valve abnormalities), delayed
puberty, characteristic facial features (such as short broad neck with
webbing, micrognathia, low set ears and down-slanted palpebral fissures
with epicanthal folds), or infertility
If 2 or more features commonly associated with
Turner syndrome such as renal anomaly (hypoplasia, aplasia
or horseshoe kidney), other cardiac anomalies (e.g., partial anomalous
pulmonary venous return, atrial or ventricular septal defects), hearing
loss, Madelung deformity, dysplastic nails, multiple nevi, and
neuropsychological issues associated with short stature are seen in a
girl
Current & Past Medical History
Girls with Turner syndrome often come to medical attention
during infancy because of the presence of distal lymphedema, nuchal redundancy,
and/or murmurs associated with characteristic left-sided cardiac malformations,
such as coarctation of the aorta and bicuspid aortic valve. Older girls may
present with unexplained short stature. Because of abnormalities in craniofacial
development, these individuals may have a history of chronic middle ear
effusions. Adolescent females with Turner syndrome may present with primary
amenorrhea. Inquire about the following:
Exercise intolerance, chest, or back pain, which may be
a symptom of aortic dilatation or impending rupture
Acute and chronic otitis media, persistent middle ear
effusion, and associated hearing loss
Problems with vision
Dental and orthodontic care, tooth abnormalities,
dental pain, and difficulty chewing
Symptoms of hyper- or hypothyroidism, which may
indicate the onset of autoimmune thyroiditis
Intercurrent UTIs, urinary frequency, and urgency
(urinary tract infections may increase the risk for chronic renal
disease and hypertension)
Abdominal pain, bloating, flatulence, chronic
constipation, or diarrhea, and poor weight gain, which may reflect
celiac disease (prevalence of 4-6% in Turner
syndrome)
Chronic abdominal pain, diarrhea, and/or constipation,
which may indicate inflammatory bowel disease
Melena (dark, tarry stools) or blood in the stool,
which may indicate the presence of an intestinal vascular
malformation
Pregnancy/Perinatal History
Fetal ultrasonography may reveal increased nuchal
translucency, cystic hygroma (the result of jugular lymphatic obstruction or
malformation), left-sided cardiac defects, renal malformation, brachycephaly,
poly- or oligohydramnios. Maternal quadruple screen may be abnormal. Maternal
quadruple serum screening may also be abnormal but, because of a high rate of
false-positives, confirmatory testing with amniocentesis or chorionic villous
sampling is necessary to entertain the diagnosis of Turner syndrome prenatally.
Karyotype should be repeated postnatally in all individuals who were previously
diagnosed prenatally. [Shankar: 2018]
Developmental & Educational Progress
Ask about school progress and relationships with family and peers. Preventive pediatric health care should include annual
developmental and behavioral screenings. Conduct a neuropsychological evaluation during early life (preschool), school entry,
transition to high school and higher education, or any time that difficulties arise. [Gravholt: 2017]
Maturationalprogress
Girls with Turner syndrome may present with pubertal delay or primary
amenorrhea due to ovarian insufficiency. A degree of normal pubertal development
may be seen prior to ovarian failure. Some girls with Turner syndrome may
achieve spontaneous menarche. Ask about pubertal changes
and menstruation in adolescents. While most girls with Turner syndrome do not go
through puberty, up to 30% will have some spontaneous pubertal development and
2-5% may become pregnant spontaneously. For those who do experience endogenous
ovarian function, discuss birth control, prevention of sexually transmitted
disease, and the pregnancy-associated risks of aortic
dissection.
Social & Family Functioning
Inquire about family support as well as the child's involvement in age-appropriate sports and activities.
Physical Exam
Vital Signs
Resting tachycardia may be present. Hypertension often complicates Turner
syndrome in older girls and adolescents, even those without left-sided
cardiac abnormalities. Blood pressure should be measured at least annually
and charted on height-specific growth charts. The Fourth Report on the Diagnosis, Evaluation, and Treatment of High Blood Pressure in Children and Adolescents from the National Heart, Lung, and Blood Institute has blood pressure
tables (on pages 10-11) for children and adolescents. Due to the
risk of aortic dilation and dissection, hypertension should be managed
aggressively.
Growth Parameters
Height should be plotted on a Turner Syndrome Growth Chart 2-19 Years ( 1.2 MB) beginning at 2 years
of age. Growth charts for girls younger than 2 with Turner syndrome have not
been developed; in girls younger than 2, height velocity should be monitored
on a standard growth chart. Weight and body-mass index (BMI) should be
plotted on a standardized BMI chart such as BMI Females 2-20 Years (CDC) ( 68 KB), since no BMI charts for Turner
syndrome have been developed.
Skin
Dermatologic evaluation may reveal multiple hyperpigmented nevi throughout the body, which are not thought to be at increased
risk for melanoma or other skin neoplasms. In older adolescents and adults, examine the intertriginous areas for acanthosis
nigricans, a sign of insulin resistance.
HEENT/Oral
Examine for:
Bitemporal narrowing
Epicanthal folds and ptosis
Strabismus
Low-set, posteriorly rotated, prominent auricles or
attached earlobes
Chronic middle-ear effusion
Micrognathia (small mandible)
High, arched palate due to narrow maxilla
Low posterior hairline
Pterygium coli (webbed posterior neck)
Broad, short appearing neck
Ocular abnormalities are common and include
near-sightedness, far-sightedness, strabismus, ptosis, epicanthal folds, and
hypertelorism. Girls with Turner syndrome are at increased risk for
recurrent ear infections, persistent middle ear effusions, and conductive or
sensorineural hearing loss. Inspect tympanic membrane with pneumatic
otoscopy to monitor for middle ear fluid. Assess for tooth abnormalities and
malocclusion.
Chest
Visual inspection may reveal a “shield-shaped” chest with pectus excavatum and widely spaced nipples. Nipples may be hypoplastic
and/or inverted.
Heart
A systolic murmur may indicate a left-sided cardiac malformation such as bicuspid
aortic valve or aortic coarctation, which may be best heard over the left
scapula or in the axilla. Some individuals with these lesions have no
auscultatory findings; echocardiography is recommended in all cases of known
or suspected Turner syndrome. Absent or weak lower extremity peripheral
pulses may indicate aortic coarctation, and an experienced examiner may be
able to perceive brachiofemoral delay of the pulses.
Abdomen
Palpate the abdomen for a mass that may signify a renal abnormality such as collecting system malformation with obstruction
or horseshoe kidney. Gastrointestinal vascular malformations may present with rectal bleeding and have been reported in individuals
4 months old to 57 years old.
Genitalia
Follow Tanner stage and consider referral for pubertal induction if no pubertal development has occurred by 12 years of age.
Extremities/Musculoskeletal
Examine for cubitus valgus, short 4th metacarpal bones, Madelung deformity of the wrist, scoliosis/kyphosis, and genu valgum
or genu varum. Developmental dislocation of the hip can occur. Radiography may reveal a coarse trabecular pattern, particular
at the metaphyses of long bones. Congenital lymphedema may result in residual puffiness of the dorsae of the hands and feet.
Fingernails and toenails may be narrow, hyperconvex, and/or deep-set.
Testing
Sensory Testing
An audiologist should perform a hearing evaluation at diagnosis and every
3-5 years thereafter. If there is a history of otitis media or hearing loss,
evaluations are usually performed annually. Turner
syndrome is associated with red-green color blindness (10%), hyperopia (35%),
and strabismus (25%) with risk of amblyopia. Girls with Turner syndrome should
be evaluated by a pediatric ophthalmologist by 12-18 months of age or earlier if
clinically indicated. If initial evaluation is normal, the medical home provider
should conduct annual routine vision screening.
Laboratory Testing
Routine laboratory studies to monitor for comorbid conditions should begin in early childhood or at the time of diagnosis. If initial diagnosis occurs before 10 years of age:
Assess thyroid function with a TSH and T4.
Screen for celiac disease starting at age 2 with a tissue transglutaminase IgA (TTG) and total serum IgA.
If initial diagnosis occurs at 10 years of age or later:
Assess thyroid function with a TSH and T4.
Screen for celiac disease with a tissue transglutaminase IgA (TTG) and total serum IgA.
Perform hepatic function panel, GGT, HbA1c with or without fasting plasma glucose, fasting lipid panel.
25-hydroxyvitamin D
Ongoing assessment during childhood:
Hepatic function panel annually
Thyroid screening annually with a thyroid-stimulating hormone (TSH) and thyroid hormone (T4)
Celiac screen every 2 years with a tissue-transglutaminase IgA (TTG-IgA) and a total IgA
25-hydroxyvitamin D every 2-3 years
Ongoing assessment for adults:
Fasting lipid panel and HbA1c with or without fasting plasma glucose annually
Hepatic function panel annually
Thyroid screen with TSH and T4 annually
Celiac screen with TTG-IgA and total IgA with suggestive symptoms
Echocardiography and cardiac MRI should be performed on all
individuals with Turner syndrome (50% of girls with Turner syndrome have
congenital heart malformations). [Lin: 2008] Prenatal
detection of Turner syndrome should prompt a fetal echocardiogram and referral
to pediatric cardiology. Surveillance for aortic root dilation, treatment for
hypertension, prophylactic medical therapy, and surgical consultation when
appropriate are essential to reduce the incidence of aortic dissection.
[Gravholt: 2006] Cardiac imaging may be performed
with 2-dimensional and color Doppler echocardiography if performed along with a
baseline electrocardiogram with QTc measurement and evaluation from a pediatric
cardiologist. Echocardiography is generally sufficient in infants and young
children, but thoracic abnormalities and obesity may limit its use in older
individuals. [Bondy: 2007]
If echocardiography is inadequate, computed tomography
or cardiac magnetic resonance imaging should be performed at a center with
expertise. Whichever imaging modality is used, it is imperative that the aortic
valve leaflets be adequately visualized, along with the aortic arch and
descending aorta. Depending on the cardiac malformation
present, periodic echocardiography or cardiac MRI should be performed by a
pediatric cardiologist. All individuals with Turner syndrome should undergo
cardiac MRI when they are old enough to tolerate the procedure without sedation.
Sedation may be required in younger children in whom cardiac MRI is clinically
indicated. In the absence of a bicuspid aortic valve or
other significant disease at the initial screening, TTE or cardiac MRI
surveillance studies should be performed every 5 years in children, every 10
years in adults, or prior to anticipated pregnancy. The latest consensus
guidelines assign girls less than 16 years old with Turner syndrome into low-,
moderate-, and high-risk categories based on a Turner syndrome-specific Z-score
of the aorta and recommend TTE and pediatric cardiology follow-up every 5 years,
1–2 years, and 6 months-1 year respectively. [Gravholt: 2017]
Renal ultrasound should be performed at the time of
diagnosis to identify any presence of urologic
abnormalities. DEXA scan should be done to monitor bone
health after adult hormone replacement has been initiated, every 5 years
thereafter, and at the discontinuation of estrogen therapy at
menopause.
Genetic Testing
All individuals with suspected Turner syndrome should have a standard
30-cell karyotype performed as recommended by the American College of Medical
Genetics. This identifies up to 10% mosaicism with 95% confidence. [Bondy: 2007] Additional metaphases may be counted or
fluorescence in-situ hybridization studies (FISH) may be performed if there is a
strong suspicion of undetected mosaicism. In this case, a cytogeneticist should
be consulted. A second tissue source, such as skin fibroblasts, buccal mucosa
cell, or bladder epithelial cells may be tested if clinical suspicion for
mosaicism persists despite a normal karyotype. [Wiktor: 2005]
Other Testing
Neuropsychological testing should be performed
to identify and accommodate learning disabilities in early life (preschool), at school entry, at transition to high school
and higher education, or at any time that difficulties arise.
A team of geneticists and genetic counselors should be involved in the initial diagnosis and may help guide testing, particularly
if a karyotype is normal and a suspicion for mosaicism exists. The genetics team may also coordinate initial testing for associated
conditions and counsel regarding risk for long-term complications.
Every child with Turner syndrome should be evaluated by a pediatric
cardiologist who will guide the choice of imaging study to evaluate for
cardiac abnormalities and give counsel regarding the increased risk of
atherosclerotic heart disease, aortic dilation, and aortic dissection later
in life.
Girls should be evaluated for red-green colorblindness, strabismus,
near-sightedness, far-sightedness, and hyperopia with the risk of amblyopia
by 12-18 months of age or at diagnosis in older girls with Turner syndrome
and then annually thereafter.
Referral may be helpful in ensuring optimal health monitoring, identifying comorbid conditions, assessing developmental progress,
ensuring optical intervention services, and managing behavioral concerns such as attention-deficit disorder.
A team (including a developmental pediatrician, psychologist, neurologist, speech, and occupational therapist) will conduct
an interdisciplinary assessment of developmental and functional abilities for girls with persistent challenges in learning,
attention, or behavior.
Testing should be considered at major transitional stages (preschool, entry to elementary, and high school).
Treatment & Management
Overview
Managing Turner syndrome can be challenging because of the constellation of
potential abnormalities. Patients with Turner syndrome should ideally be managed in
centers with pediatric sub-specialists.
Pearls & Alerts for Treatment &
Management
Pregnancy is possible and may come with high risks
Pregnancy in individuals with Turner syndrome is associated with rare, but
potentially fatal, aortic dissection and rupture. While gonadal failure with
pubertal delay is common, up to 30% of young women with Turner syndrome will
experience some spontaneous pubertal development and 2-5% of women will
experience spontaneous pregnancy. When appropriate, prevention of unwanted
pregnancy and potential cardiovascular complications of pregnancy should be
discussed. Women with Turner syndrome and aortic valve/aortic anomalies
should be counseled about the dangers associated with pregnancy and should
consult with cardiology so that they can make an informed decision. See the
Maturation/Sexual/Reproductive system below for more
detail.
Endocrine consult
An endocrine consult should be considered in girls with Turner who have not achieved puberty by 12 years of age.
Estrogen replacement
Turner syndrome is usually accompanied by hypergonadotropic hypogonadism and
primary or secondary amenorrhea. Most individuals with Turner syndrome will
therefore need hormonal replacement therapy initially for induction of
puberty and later for maintaining secondary sexual characteristics,
attaining optimal bone mass, normalizing uterine growth (for possible
pregnancy later). [Gravholt: 2017]
How should common problems be managed differently in children with Turner Syndrome?
Growth or Weight Gain
Height should be monitored on a Turner syndrome-specific growth chart
(see Resources/Clinical Tools/Growth/BMI Charts,
below).
Development (Cognitive, Motor, Language, Social-Emotional)
Developmental and behavioral issues are common; see Clinical Assessment/Testing, above, for details about evaluation.
Prescription Medications
A variety of electrocardiographic and repolarization
abnormalities, such as resting tachycardia, right axis deviation, T wave
abnormalities, accelerated AV conduction, and QT interval prolongation, have
been described in Turner syndrome. It is hypothesized that impaired
sympathovagal tone plays a role. Though the clinical significance of these
observations is unclear, it is recommended that individuals with QT prolongation
avoid medications that could further prolong the QT interval. [Bondy: 2007]
Systems
Endocrine/Metabolism
Short stature Short stature is the most common finding and is nearly always
present, even in patients who do not display other phenotypic
characteristics. The etiology of growth failure is poorly understood, but it
is thought to involve skeletal dysplasia and poor responsiveness to growth
hormone related to haplo-insufficiency for the short stature
homeobox-containing (SHOX) gene on the X-chromosome.
Growth hormone studies in these patients are typically normal. During
infancy and childhood, growth rates are approximately 2 standard deviations
below mean growth rates. The adult height of girls not treated with growth
hormone is 56 to 57 inches (about 8 inches below the average adult woman's
height). Growth hormone secretion pattern is usually normal. Recombinant
growth hormone (GH) therapy has been shown to improve adult height in
patients with Turner syndrome by 5–8 cm in several studies, but the efficacy
is variable and depends on multiple factors including age at initiation,
baseline heights, genetic potential, and dose and duration of therapy. Growth hormone (GH) therapy Initial consultation with a pediatric endocrinologist should
determine the appropriate timing for beginning therapy. The recent
guidelines recommend initiating GH treatment early (around 4–6 years of age
and preferably before 12–13 years) in the following circumstances:
The child already has evidence of growth failure
(e.g., below 50th percentile height velocity observed over 6 months
in the absence of other treatable causes of poor
growth).
The child is already short or has a strong
likelihood of short stature (e.g., short parents and short predicted
adult height or already pubertal at the time of
diagnosis).
The recommended GH dose per the recent guidelines is
45–50µg/kg/day in most instances, increasing to 68µg/kg/day (2.0mg/m2/day)
if initial response is suboptimal. Height should be monitored every 4-6
months during the first year of treatment and every 6 months thereafter.
Therapy with GH is generally well tolerated, although there is a slightly
higher risk of serious adverse effects such as intracranial hypertension and
slipped capital femoral epiphysis. [Bolar: 2008]
Although human growth hormone (hGH) does not appear to increase the risk of
cancer, it is not recommended for children with active neoplastic processes.
hGH should be used with caution after renal transplant and should not be
used in individuals with closed epiphyses. [Cave: 2003] See Clinical Practice Guidelines for Growth Hormone Use in Adults and Children (AACE) ( 44 KB). IGF-1 levels should be monitored at least yearly to monitor safety of GH
therapy. The measured IGF-1 should ideally be no greater than 2SDs above the
mean for age. If the IGF-1 is above +3SDs, a GH dose decrease is warranted. Oxandrolone In girls with Turner syndrome older than 10 years of age with
poor projected adult height on GH alone, the addition of oxandrolone, an
androgen and anabolic steroid, at 0.03-0.05 mg/kg/day may be considered.
[Perry: 2014]
[Gravholt: 2017] Oxandrolone may improve adult
height by 2-5 cm when used with GH. [Menke: 2010]
Potential side effects are less of a concern at the low dosages above, but
they may include acne and clitoromegaly. [Perry: 2014]
Autoimmune dysfunction Autoimmune thyroiditis is common and may be seen as young as
4 years of age. One longitudinal study found that 24% of children over 8
years old with Turner syndrome developed hypothyroidism and 2.5% developed
hyperthyroidism. [Livadas: 2005] Because clinical
symptoms of thyroid dysfunction rarely manifest, annual screening with FT4
and TSH is recommended starting in early childhood and annually throughout
the lifespan. Thyroid replacement should be prescribed in those with
hypothyroidism. Diabetes The risk of both type 1 and type 2 diabetes mellitus is about
a 10-fold and 4-fold increase in patients with Turner syndrome across all
ages in epidemiological studies. Obesity, insulin resistance, and impaired
glucose tolerance are also common. Lifelong annual
measurement of HbA1c, with or without fasting plasma glucose, is recommended
starting at age 10 years. If testing meets criteria for diabetes, the
patient should be assessed for antibodies related to type 1 diabetes and be
seen by a diabetes specialist. Dyslipidemia Dyslipidemia has been reported as young as 11 years and is
independent of BMI. Nutrition and exercise counseling are an important
component of ongoing care. Regular moderate exercise should be encouraged.
Hypercholesterolemia has been reported in 37–50% of women with Turner
syndrome. [Garden: 1996]
According to the American Academy of Pediatrics and
American Heart Association guidelines, non-fasting, non-HDL cholesterol
(calculated by subtracting HDL cholesterol from total cholesterol) should be
measured on 2 occasions: once between 9 and 11 years old and again between
17 and 21 years prior to transition to adult care. If non-HDL cholesterol is
≥145mg/ dL (≥3.7mmol/L), then a full fasting lipid profile should be
obtained. A lipid profile should be performed in individuals who have at
least 1 risk factor for cardiovascular disease starting at age 18 years.
[Gravholt: 2017]
An endocrinologist will monitor growth velocity, guide the decision to
initiate growth hormone therapy, monitor for associated adverse
effects, guide the timing and medical management of pubertal
induction, and assist in the diagnosis and treatment of Turner
syndrome-associated hypothyroidism, obesity, and insulin
resistance.
Maturation/Sexual/Reproductive
Pubertal induction Ovarian insufficiency is a hallmark feature of Turner
syndrome. While up to 30% of girls with Turner syndrome have some
spontaneous pubertal development, gonadal failure is more common. In many
individuals, pubertal delay plays a large role in self-esteem, anxiety, and
social isolation. In the past, pubertal induction was delayed until 15 years
of age to maximize height potential. Age-appropriate pubertal induction is
now recommended to avoid the potential long-lasting psychosocial effects of
delayed pubertal development. Pubertal induction should be performed in
consultation with an endocrinologist. Serum gonadotropins (especially FSH)
should be assessed annually starting at about 11 years, prior to pubertal
induction, to exclude the possibility of impending delayed spontaneous
puberty. Low levels of anti-Müllerian hormone (AMH) and inhibin B
measurements have also been shown to predict ovarian insufficiency, and AMH
is perhaps the best indicator of ovarian reserve. If gonadotropins are
normal for age, observation for spontaneous puberty is appropriate with
future replacement therapy if gonadal failure occurs.
Transdermal 17-β estradiol (TDE) is now the
preferred treatment starting around age 11–12 years. It is a more
physiologic mode of delivery than oral estrogen and has better
bioavailability. Replacement is usually initiated at one-tenth to one-eighth
of the adult replacement dose and gradually increased over 2-4 years.
Progestin supplementation should be started once withdrawal bleeding is
noted or after about 2 years of estrogen therapy to minimize the risk of
endometrial cancer due to unopposed estrogen effect. The use of oral
contraceptive pills to induce puberty is not recommended because the
synthetic estrogen doses are higher than the desired physiologic doses and
synthetic progestin may interfere with optimal breast and uterine
development. Routine supplementation of very low-dose estrogen in childhood
to improve growth or bone mass is currently not recommended. [Shankar: 2018]
Pregnancy As the patient with Turner syndrome matures, it is important
to engage her in discussions about how Turner syndrome and its treatment
affect sexual development, function, and reproductive potential. While most
women are infertile, 2-5% may become pregnant. Others may achieve pregnancy
through various reproductive assistance techniques. Young mosaic Turner
syndrome women with persistent ovarian function should be counseled that
oocyte cryopreservation after controlled ovarian hyperstimulation is a
possible fertility preservation option. Pregnancy in Turner syndrome is associated with the rare but
potentially fatal complication of aortic dissection and
rupture. Any woman with Turner syndrome considering pregnancy
should have a cardiac evaluation. Other Turner syndrome-related pregnancy
complications include hypertension, gestational diabetes, and need for
caesarian section due to small maternal size. When appropriate, prevention
of sexually transmitted disease and unwanted pregnancy should be addressed.
[Bondy: 2007]
Women with abnormalities of the aortic valve or aorta should be
should be counseled about the dangers associated with pregnancy and
should consult with cardiology so that they can make an informed
decision. Pregnancy is considered high risk in women with
Turner syndrome who have an ascending ASI >2–2.5 cm/m2 due to risk of
aortic dissection; assisted reproductive techniques are contraindicated. If
pregnancy occurs, it should be managed with strict treatment of
pregnancy-associated hypertension, frequent cardiac imaging, and
consideration of prophylactic surgery if rapid aortic enlargement is seen.
[Shankar: 2018]
Neoplasms The presence of Y-chromosome material is associated with an
approximately 10% risk of gonadoblastoma. [Mazzanti: 2005]
[Cools: 2006] Prophylactic removal of gonadal
streaks in these individuals is recommended.
A delivery plan should be made by a multidisciplinary team consisting of at least an obstetrician, cardiologist, and anesthesiologist
that all have expertise in pregnancy in the context of maternal heart disease and/or aortopathy.
Cardiology
Cardiovascular malformation The most serious conditions associated with Turner syndrome
involve the cardiovascular system. Congenital heart disease occurs in
approximately 22-70% of women with Turner syndrome. [Mortensen: 2012] While left-sided cardiac anomalies are most common,
the wider range of malformations includes aortic coarctation (11%), bicuspid
aortic valve (15%), partial anomalous pulmonary connection (13%), persistent
left superior vena cava (13%), mitral valve abnormalities (<5%), and
rarely hypoplastic left heart syndrome. Coarctation may not be detected on
echocardiography during infancy but found with the first cardiac MRI.
Generalized dilation of major vessels has been described, although the
clinical significance of this is unclear. [Lin: 2008]
Individuals with Turner syndrome are at risk for rare but
potentially fatal aortic dilation, dissection, and rupture, even in
relatively young individuals. The risk for dissecting aortic
aneurysm is greater in those with aortic valve abnormalities,
coarctation/dilation of the aorta, and systemic hypertension. Counsel
at-risk patients and their families about the need for medical monitoring
and treatment and the potential symptoms of aortic dissection (chest or back
pain). They should also be encouraged to carry medical information at all
times to alert medical personnel to the presence of aortic disease. The risk
of aortic dissection with pregnancy should be discussed at length with those
who have endogenous ovarian function and are considering assisted pregnancy.
Those with CHD diagnosed in childhood may be at risk
for postoperative valve re-stenosis, aortic re-coarctation, or residual
septal defect shunts and should be monitored closely for the development of
new or changing cardiac murmurs. There is a strong association
between neck webbing and the presence of a congenital heart
defect. Resting tachycardia and a variety of
electrocardiographic and repolarization abnormalities have been recognized
in Turner syndrome, including prolongation of the QT interval.
The latest consensus guidelines assign girls with
Turner syndrome aged <16 years into low-, moderate-, and high-risk
categories based on a Turner syndrome-specific Z-score of the aorta and
recommend transthoracic echocardiogram (TTE) and pediatric cardiology
follow-up every 5 years, 1–2 years, and 6 months-1 year, respectively. In
individuals with Turner syndrome over the age of 16 years, the ascending
aortic size index (ASI), defined as the aortic diameter in cm corrected for
body surface area, is a useful prognostic indicator and, has been used to
categorize risk (2–2.3 cm/m2 is moderate risk and >2.3 cm/m2 is high
risk) and suggest therapy in the latest guidelines. [Gravholt: 2017]
[Shankar: 2018]
Eligibility for competitive sports for individuals
with Turner syndrome should be determined by a cardiologist after a
comprehensive evaluation. Participation in sports is restricted to low and
moderate static and dynamic activities in the moderate risk category while
girls in the high-risk category should avoid competitive sports and intense
weight training. [Płytycz: 1986]
For patients with no cardiovascular malformation,
routine pediatric care should include annual measurement of blood pressure.
In the absence of a bicuspid aortic valve or other significant diseases at
the initial screening, TTE or cardiac magnetic resonance (CMR) surveillance
studies should be performed every 5 years in children, every 5-10 years in
adults, or prior to anticipated pregnancy. [Gravholt: 2017]
[Bondy: 2007]
The importance of regular moderate exercise should
be stressed. Intense or contact activities, as well as isometric exercises,
may unnecessarily stress the cardiovascular system. Hypertension Half of women with Turner syndrome have hypertension.
Although the exact etiology remains unclear, it is possibly due to
small-vessel renovascular disease. [Ostberg: 2003]
One case series identified coarctation or renal disease as primary causes of
hypertension in only 20% of hypertensive Turner syndrome women. [Elsheikh: 2002] Hypertension should be treated
aggressively. In individuals without structural heart disease, annual
assessment of blood pressure should be performed and medical treatment
should be considered if hypertension is present. Medical treatment would
include a beta-blocker, an angiotensin receptor blocker, or both to reduce
the risk for aortic dissection in women with Turner syndrome who are ≥16
years of age for whom their ascending ASI is ≥2.3cm/m2. [Gravholt: 2017]
Electrocardiographic abnormalities Every individual with Turner syndrome should receive a
resting electrocardiogram (ECG) with QTc measurement at diagnosis.
Electrocardiographic and repolarization abnormalities, such as resting
tachycardia, right axis deviation, T-wave abnormalities, accelerated AV
conduction, and QT-interval prolongation have been described in Turner
syndrome. Some changes such as those in P-wave and QTc-dispersion and
heart-rate variability in women with Turner syndrome can be attributed to
the underlying characteristic autonomic dysfunction. The clinical
significance of these observations is unclear. It is recommended, however,
that individuals with QT prolongation avoid medications that could further
prolong the QT interval. [Bondy: 2007] If they are
deemed necessary, ECG should be performed 1–2 weeks after initiation of
QT-prolonging drugs. Coronary artery disease Coronary artery disease is thought to be twice as common in
women with Turner syndrome as in the general population. Risk factors
include hypertension, insulin resistance, dyslipidemia, and estrogen
deficiency. [Ostberg: 2003]
A pediatric cardiologist will closely follow the child with Turner
syndrome, make recommendations for management of any cardiac
malformations, and assist the medical home provider in managing
hypertension.
Ears/Hearing
Individuals with Turner syndrome often have a flattened cranial base angle
resulting in an abnormal relationship between the Eustachian tube and the
middle ear. This can lead to a high prevalence of otitis media, persistent
middle ear effusion, and increased risk for conductive hearing loss. Those
with persistent otorrhea are at risk for cholesteatoma formation.
Sensorineural hearing loss is also prevalent; while the onset of this
comorbidity is typically in adulthood, it has been described in patients as
young as 6 years old and may necessitate the use of amplification.
Perform (at least) annual evaluations for middle ear
effusions that include pneumatic otoscopy and tympanometry. Otitis media
should be treated aggressively because of the significant impact that
hearing loss can have on speech and language development and the risk of
cholesteatoma formation in those with persistent otorrhea. Persistent middle
ear effusion, particularly if associated with language delay or ongoing
symptoms of illness, should prompt referral to an otolaryngologist to
consider placement of pressure equalization tubes and perhaps tonsillectomy
and/or adenoidectomy. Adenoidectomy may, however, exacerbate palatal
dysfunction negatively impacting speech quality and should be considered
with caution. See the Portal's Hearing Loss and Deafness for management
information.
Refer for diagnosis and every 1-5 years thereafter as recommended or as indicated by subjective hearing changes, persistent
middle ear effusions, or recurring suppurative otitis media.
Referral to an otolaryngologist should be considered for persistent middle ear fluid lasting longer than 3 months or for recurrent
suppurative otitis media.
Eyes/Vision
Strabismus occurs in 25% of girls with Turner syndrome, hyperopia in 35%, and
red-green colorblindness in 10%. Abnormalities of the external ocular
adnexa, such as epicanthal folds and ptosis, are common. Cataracts and
nystagmus occur more commonly in Turner syndrome as well. Infants should be
screened carefully for strabismus, and all should be evaluated by an
ophthalmologist by 12-18 months of age or at diagnosis in older girls and
annually thereafter.
Referral should be considered at diagnosis or by 18 months of age. Follow-up will depend on whether and which abnormalities
are identified.
Dental
Abnormal tooth eruption and root and crown abnormalities may occur. A flattened cranial base angle, decreased posterior cranial
base length, and retrognathia may lead to dental malocclusion and bite abnormalities. Treatment with growth hormone can alter
craniofacial proportions leading to further orthodontic concerns. These patients are also at risk for abnormalities in tooth
development and morphology, such as early eruption of secondary teeth, simple crown morphology, thin/hypoplastic dental enamel,
short dental roots, and root resorption leading to tooth loss. Patients with cardiac abnormalities should be considered for antibiotic prophylaxis for subacute bacterial endocarditis prior
to dental procedures.
Girls with a narrow maxilla and a relatively wide mandible have a high prevalence of malocclusion and should see a pediatric
orthodontist no later than 7 years of age and have regular orthodontic follow-up. Growth hormone therapy can alter the craniofacial
proportions as well.
It is recommended that individuals with Turner syndrome see a pediatric
dentist by 2 years of age and have regular follow-up at intervals
determined by the problems identified.
Musculoskeletal
Scoliosis and kyphosis Monitor annually for scoliosis during routine pediatric care
or every 6 months if on growth hormone therapy. Spinal curvature may
progress with rapid growth. While current literature suggests that growth
hormone does not cause scoliosis, it may accelerate the development of a
spinal curve. Evaluation by an orthopedist is recommended in cases of
significant spinal curvature or in those whose curvature is detected at a
young age. Management of scoliosis (e.g., bracing or surgical intervention)
does not differ from that in the general population. Other muscular
complications could include pectus deformities. Madelung deformity Madelung deformity results from epiphyseal arrest on ulnar
and volar aspects of the distal radius causing the articular surface to be
directed toward the ulna and volar aspect of the wrist. This may result in
wrist pain as well as limited wrist extension and supination. Hip dislocation Developmental dysplasia of the hip occurs with increased
frequency. Conservative management with bracing (e.g., a Pavlik harness) or
casting is recommended as in the general population with surgical
intervention reserved for dysplasia not responsive to bracing. Cubitus valgus Increased cubital carrying angle may limit range of motion
and interfere with function. Chronic knee pain An abnormal tibial plateau coupled with patellar changes may
lead to chronic knee pain. Decreased bone mineral density Decreased bone mineral density (BMD) with increased fracture
risk can occur in older individuals, particularly those not treated with
estrogen. Short stature may, however, lead to an underestimation of
bone-mineral density by dual-energy x-ray absorptiometry (DEXA). When
adjusted for height, women with Turner syndrome who receive appropriate
estrogen therapy have an estrogen-independent decrease in cortical BMD with
normal trabecular BMD. Causes of low BMD may include non-adherence to
estrogen therapy, tobacco use, excessive alcohol use, celiac disease, and/or
vitamin D deficiency. The importance of weight-bearing exercise in reaching
and maintaining adequate BMD should be discussed with patients and families.
Bisphosphonates are not recommended in young women because the reduced
cortical BMD is not associated with an increased risk of fractures, and
these medications have not been shown to increase cortical BMD.
Bisphosphonate use may be considered in women with confirmed osteoporosis,
those at risk for fracture, and those who have sustained a low-impact
fracture. [Bondy: 2007]
Screen for vitamin D deficiency with a 25-OH vitamin
D concentration between 9–11 years and every 2–3 years thereafter. DEXA
scans should be obtained to monitor bone health after adult hormone
replacement has been initiated, every 5 years thereafter, and at
discontinuation of estrogen therapy at menopause. See Osteoporosis and Pathologic Fractures.
Refer for diagnosis of scoliosis, hip dysplasia, and range of motion abnormalities associated with skeletal dysplasia. Orthopedics
will recommend optimal management (e.g., bracing vs. surgical correction) and assist in the management of functional limitations
caused by skeletal dysplasia (e.g., limited elbow extension or pain related to cubitus valgus).
Refer for evaluation and assistance in management of osteopenia.
Renal
Malformations of the urinary system occur in 30-40% of individuals with
Turner syndrome. Collecting system abnormalities and abnormal positioning
are seen most frequently (15-20%), followed by horseshoe kidneys (10%) and
aplasia (3%). If structural renal malformations are detected on initial
ultrasonography, follow-up and treatment plans will be individualized based
on the condition and the recommendations of the involved subspecialists. The
majority of renal malformations seen in Turner syndrome do not result in
renal dysfunction or disease. In some, however, complications such as
hydronephrosis, reflux, and/or recurrent urinary tract infections may be
severe and require treatment and ongoing monitoring. In patients with no
underlying urinary tract malformation, there is no increased risk of
developing renal disease. [Bondy: 2007]
Refer for help in evaluating and managing renal or collecting system malformations and related problems.
Skin & Appearance
Lymphedema Lymphedema is a major feature of Turner syndrome. The
etiology is uncertain, but it is thought to arise from a generalized
lymphatic dysplasia. It results in ongoing puffiness of the hands and feet,
as well as nuchal webbing, the classic appearance of the chest and atypical
configuration of the finger and toenails. Palmar and pedal edema may be
exacerbated by estrogen and growth hormone therapy. Symptomatic relief of
edema with support socks, elevation, or compression dressings may be
required. Chronic diuretic therapy is not recommended as it is marginally
effective and may lead to fluid and electrolyte imbalances. Vascular surgery
should be avoided. [Bondy: 2007] Patients and
families can be directed to The National Lymphedema Network for more information about
lymphedema and its management. Melanocytic nevi Individuals with Turner syndrome have an increased number of
typical melanocytic nevi. Studies conflict about whether Turner syndrome is
associated with increased melanoma risk. Therapy with GH may trigger
melanocyte growth, but it has been shown neither to increase the number of
nevi nor to trigger malignant transformation. Other skin conditions that
have a greater prevalence include pilomatricomas, vitiligo, and halo nevi.
Nevi should be monitored for concerning changes in
shape, color, and size. It has been suggested that girls and women with
Turner syndrome are at increased risk for keloid formation as well, although
it is unclear whether this is due to an abnormal healing process or whether
this is related to surgeries often involving the head and neck - areas more
likely to exhibit keloid formation.
Refer for surgical management of undesirable scars and nevi.
Gastro-Intestinal & Bowel Function
Gastroesophageal reflux Feeding problems during infancy are relatively common and may
include difficulties latching and sucking, gastroesophageal reflux, and
failure to thrive. Anatomical differences in the oropharynx (high palate,
palatal insufficiency), oral-motor immaturity, and abnormal gastrointestinal
motility may contribute to these problems. Gastroesophageal reflux may
safely be treated with H2 receptor blockade or proton pump inhibitors.
Nasogastric tube feedings may be required if feeding difficulties are
severe. Referral to a speech or occupational therapist for a feeding
evaluation may be helpful. In some cases, simple interventions such as
elevation of the head after feeds and use of a specialized nipple may result
in improvement. [Cassidy: 2005] See the Medical
Home Portal's Gastroesophageal Reflux Disease for more information. Inflammatory bowel disease Inflammatory bowel disease is 2-3 times more common in Turner
syndrome than in the general population, and it has a higher rate of
complications when it occurs. Colectomy has been reported in 40% of Turner
syndrome-associated inflammatory bowel disease. Other potential
complications include fistula formation and sepsis. [Ostberg: 2003] Treatment focuses on nutritional therapy and treatment
with anti-inflammatory or immunomodulatory medications. Celiac disease Celiac disease affects 4-6% of individuals with Turner
syndrome. Celiac disease refers to immune-mediated small bowel inflammation
triggered by exposure to the gliadin peptide contained in gluten and found
in wheat, barley, rye, and possibly oats. Treatment of celiac disease
consists of complete elimination of gluten from the diet and is monitored by
clinical response (including growth response), serologic marker response,
and small intestinal mucosa histologic response. See the Medical Home
Portal's Celiac Disease for
more information. Liver dysfunction Asymptomatic liver test (ALT, AST, and GGT) abnormalities are
a common finding with increasing prevalence with age (20–80%) and an annual
incidence of 2.1–3.4%. Liver enzyme elevations tend to persist or
progressively increase and rarely revert to
normal. Liver function should be assessed at age 10
and annually thereafter or as clinically indicated. If elevated hepatic
enzymes persist for more than 6-12 months, liver ultrasound should be
performed to evaluate for hepatic steatosis. If steatosis is not present but
elevated hepatic enzymes persist, a hepatology consult should be obtained.
Potentially, hepatotoxic medications such as statins and glitiazones should
be prescribed with caution in patients with hepatic enzyme elevation.
[Bondy: 2007] Structural changes such as fatty
infiltration, fibrosis, vascular changes, and nodular hyperplasia have been
reported on liver biopsy, but their relationship to liver enzyme elevation
has not been defined. The mechanism of liver disease
in Turner syndrome is not well understood, but it is thought to be
multifactorial, with obesity and metabolic syndrome as likely contributors.
Other possible risk factors include vascular anomalies, as evidenced by
nodular regenerative changes, biliary lesions (e.g., primary sclerosing
cholangitis) and autoimmunity (e.g., primary biliary cirrhosis). Several
studies have now documented improvement or resolution of liver enzyme
elevation with estrogen replacement therapy. Intestinal telangiectasia Patients with Turner syndrome are at risk for vascular
malformations throughout the gastrointestinal tract, which may present with
rectal bleeding. Refer to a gastroenterologist for endoscopy and management
for persistent rectal bleeding or melanotic stools. Colonic carcinoma A few small studies have indicated an increased relative risk
of colon cancer in people with Turner syndrome. Current recommendations for
colon cancer screening (or treatment) in Turner syndrome are no different
from those in the general population.
May assist in the interpretation of antibody tests in the diagnosis of celiac disease or perform confirmatory testing through
endoscopy with biopsy. Consider consultation for persistently elevated hepatic enzymes, chronic abdominal pain, constipation,
diarrhea, rectal bleeding, or melanotic stools.
Referral to speech or occupational therapy for a feeding evaluation may be helpful.
Development (general)
Ideally, a neuropsychological evaluation should be conducted during early
life (preschool), school entry, transition to high school and higher
education, or at any time that difficulties arise. If a neuropsychologist
(or otherwise qualified psychologist) is not a member of the
multidisciplinary team, then direct efforts at identifying community
providers who can provide needed evaluations (e.g., school psychologists).
It is also recommended that children be referred for occupational, physical,
and speech therapy in early life or at school entry as warranted.
Most girls with Turner syndrome will have normal
motor, cognitive, and language development, although up to 10% may have
developmental delay requiring early intervention or special education.
[Sybert: 2004] As girls with Turner syndrome
reach school age, a common neurocognitive profile includes lower scores on
nonverbal than verbal performance tests. This may result in difficulties
with nonverbal abilities, such as math and visual-spatial skills, or
difficulties with executive functioning and slower processing speed. These
girls may benefit from accommodations in academic testing situations.
Despite variable learning difficulties, girls and women with Turner syndrome
often have excellent verbal skills and many achieve college-level
education.
Referral may help with monitoring health, identifying comorbid conditions, assessing developmental progress, ensuring optical
intervention services, and managing behavioral concerns such as attention-deficit disorder.
Mental Health/Behavior
Individuals with Turner syndrome may have delayed emotional maturation, poor
relations with peers, timidity, and negative body image. For those with
difficulties in executive functioning, consider referral to a social-skills
group or class. Early psychoeducational testing with appropriate classroom
supports will help an affected child succeed academically, reducing the risk
for psychosocial problems related to poor school performance. In older
girls/adolescents, poor self-esteem may be related to delayed puberty, and
pubertal induction is recommended at 12 years of age if no spontaneous
pubertal development has occurred. [Loscalzo: 2008]
Attention-deficit disorder is relatively common in girls with Turner
syndrome. [Loscalzo: 2008] Timely, comprehensive
psycho-educational evaluations with re-evaluations as needed,
age-appropriate pubertal induction, peer engagement, and career and
vocational planning for the best long-term outcomes are
recommended.
Evaluation by a learning specialist may help identify specific learning disabilities and plan educational strategies.
Transitions
The pediatric endocrinologist (or any other care provider/coordinator) should
implement a planned and staged transition process in early adolescence for
their patients with Turner syndrome. It is recommended that pediatric
practices use Turner syndrome-specific transition toolkits such as Tools for Clinicians Transitioning Women with Turner Syndrome (ACP) ( 410 KB). Turner syndrome patients will need ongoing surveillance for
co-morbidities, including Type 2 diabetes mellitus, fatty liver,
sensorineural hearing loss, hyperlipidemia, and hypertension. Surveillance
for aortic root dilation and treatment of other cardiac abnormalities by a
cardiologist familiar with the care of adults with congenital heart diseases
is important. A baseline DEXA scan is recommended after adult hormone
replacement has been initiated, and follow-up scans should be obtained every
5 years and at discontinuation of estrogen therapy at menopause. Estrogen
supplementation should ideally be continued until menopausal age to optimize
bone health and prevent osteoporosis in Turner syndrome patients.
[Shankar: 2018]
Specialty Collaborations & Other Services
Care for adults with Turner syndrome is ideally delivered in a
multidisciplinary setting with endocrinology, gynecology, cardiology,
and gastroenterology as needed among other specialties. Specialized Centers of Turner Syndrome Care (TSF) lists such clinics in several states.
Ask the Specialist
When should a child with Turner syndrome begin therapy with growth hormone?
The recent guidelines recommend initiating GH treatment early (around 4–6 years of age and preferably before 12–13 years)
if the child already has evidence of growth failure (e.g., below 50th percentile height velocity observed over 6 months in
the absence of other treatable cause of poor growth) or if the child is already short or has a strong likelihood of short
stature (e.g., short parents and short predicted adult height or already pubertal at the time of diagnosis).
When should an adolescent with Turner syndrome undergo induction of puberty?
In the past, medical induction of puberty with estrogen was delayed until about 15 years of age to maximize a patient's height
potential. Recent data indicates that beginning pubertal induction with estradiol at age 11-12 years allows a normal pace
of puberty without interfering with the effect of growth hormone on ultimate height. Delaying pubertal induction may have
several deleterious effects, including reduced bone mineralization as well as the negative psychosocial consequences of late
pubertal development. [Bondy: 2007] Gonadotropins (esp. FSH) should be monitored annually starting at about 11 years to confirm hypergonadotropic hypogonadism
prior to pubertal induction. Transdermal 17-beta estradiol is now the preferred treatment starting at around age 11-12 years.
[Gravholt: 2017]
Can women with Turner syndrome become pregnant?
While the majority of women with Turner syndrome are infertile, spontaneous
pregnancy has been reported. Some women with Turner syndrome become pregnant
through assisted reproductive technology. Reports of fatal aortic dissection
during pregnancy and the postpartum period have raised concerns about the safety
of pregnancy in Turner syndrome. If pregnancy is being considered, a full,
preconception cardiac evaluation, including an MRI of the aorta, should be
performed. Women who have a repaired cardiovascular defects, bicuspid aortic
valve, current aortic dilatation, or systemic hypertension should probably not
become pregnant. [Bondy: 2007]
What is the risk of Turner Syndrome recurring in future pregnancies?
The likelihood of a woman having a second child with Turner syndrome is no greater
than in the general population.
Resources for Clinicians
On the Web
The following modules/pages within the Portal provide more detailed
information about conditions associated with Turner syndrome:
SHOX Deficiency Disorders (GeneReviews) Detailed information addressing clinical characteristics, diagnosis/testing, management, genetic counseling, and molecular
pathogenesis; from the University of Washington and the National Library of Medicine.
Turner Syndrome (GARD) Includes information about symptoms, inheritance, diagnosis, finding a specialist, related diseases, and support organizations;
Genetic and Rare Diseases Information Center of the National Center for Advancing Translational Sciences.
Shankar RK, Backeljauw PF. Current best practice in the management of Turner syndrome. Ther Adv Endocrinol Metab.
2018;9(1):33-40.
PubMed abstract / Full Text
Klein KO, Rosenfield RL, Santen RJ, Gawlik AM, Backeljauw PF, Gravholt CH, Sas TCJ, Mauras N. Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. J Clin Endocrinol Metab.
2018;103(5):1790-1803.
PubMed abstract
Levitsky LL, Luria AH, Hayes FJ, Lin AE. Turner syndrome: update on biology and management across the life span. Curr Opin Endocrinol Diabetes Obes.
2015;22(1):65-72.
PubMed abstract
Ranke MB. Why Treat girls with Turner Syndrome with Growth Hormone? Growth and Beyond. Pediatr Endocrinol Rev.
2015;12(4):356-65.
PubMed abstract
Lee MC, Conway GS. Turner's syndrome: challenges of late diagnosis. Lancet Diabetes Endocrinol.
2014;2(4):333-8.
PubMed abstract
Milbrandt T, Thomas E. Turner syndrome. Pediatr Rev.
2013;34(9):420-1.
PubMed abstract
Turner Syndrome Health Maintenance Checklist ( 80 KB) A checklist with age recommendations for the initial and follow-up screenings of girls (birth - 18 years) with Turner syndrome;
Vana Raman, MD; based on Gravholt et al. Clinical practice guidelines for the care of girls and women with Turner syndrome:
proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017.
Turner Syndrome Growth Chart 2-19 Years ( 1.2 MB) Printable growth chart with curves for girls with and without Turner syndrome; percentiles derived from the National Center
for Health Statistics.
Laboratory Guidelines for Turner Syndrome (ACMG) Provides information on appropriate prenatal and postnatal diagnostic cytogenetic studies for Turner syndrome; American College
of Medical Genetics and Genomics.
Patient Education & Instructions
Turner Syndrome: A Guide for Families (TSSUS) ( 1.4 MB) A 32-page booklet with information for parents about growth and development, health considerations, and social and emotional
support; Turner Syndrome Society of the United States.
Turner Syndrome (Medline Plus) Information for families that includes description, frequency, causes, inheritance, other names, and additional resources;
from the National Library of Medicine.
Turner Syndrome (MedlinePlus) Information for families that includes description, frequency, causes, inheritance, other names, and additional resources;
from the National Library of Medicine.
Turner Syndrome (The Magic Foundation) Information, videos, and a variety of resources; from the Magic Foundation, a non-profit that provides support for families
of children with problems that cause problems with growth.
Turner Syndrome Foundation Supports research and facilitates education to enhance the care of those affected by Turner syndrome.
Turner Syndrome Society of the United States A non-profit with chapters and resource groups located throughout the country that provides resources to patients, families,
and physicians for the diagnosis and treatment of Turner syndrome.
Turner Syndrome Society - Local Resource Groups Links to local groups of the Turner Syndrome Society of the United States in several states, along with frequently asked questions
about the groups, what they offer, and how to develop one.
Turner Syndrome Foundation Resource Map Interactive map providing information about numbers of women with Turner syndrome in each state and how to connect with resources
via social media.
* number of provider listings may vary by how states categorize services, whether providers are listed by organization
or individual, how services are organized in the state, and other factors; Nationwide (NW) providers are generally limited
to web-based services, provider locator services, and organizations that serve children from across the nation.
Authors & Reviewers
Initial publication: September 2009; last update/revision: August 2019
Binder G, Koch A, Wajs E, Ranke MB. Nested polymerase chain reaction study of 53 cases with Turner's syndrome: is cytogenetically undetected Y mosaicism common?. J Clin Endocrinol Metab.
1995;80(12):3532-6.
PubMed abstract
Bolar K, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in turner syndrome. J Clin Endocrinol Metab.
2008;93(2):344-51.
PubMed abstract
Bondy CA. Aortic dissection in Turner syndrome. Curr Opin Cardiol.
2008;23(6):519-26.
PubMed abstract / Full Text
Bondy CA. Care of girls and women with Turner syndrome: A guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab.
2007;92(1):10-25.
PubMed abstract / Full Text This clinical practice guideline uses evidence-based data when available; expert opinion was used when evidence was lacking.
It is a comprehensive review of the diagnosis of Turner Syndrome as well as the management of associated complications.
Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. J Med Genet.
2007;44(12):745-9.
PubMed abstract
Cassidy, SB and Allanson, JE, Editors; Allanson, JE chapter author. Management of Genetic Syndromes (Noonan Syndrome Chapter). Second ed. Wiley-Liss, Inc. ;
2005.
0-471-30870-6 This chapter reviews the evaluation and management of Noonan Syndrome, pages 385-397.
Cassidy, SB and JE Allanson, Editors; VP Sybert, Chapter Author. Management of Genetic Syndromes (Turner Syndrome Chapter). Second ed. Wiley-Liss, Inc.;
2005.
0-471-30870-6 Evaluation and management of Turner Syndrome are reviewed in the Turner Syndrome chapter, pages 589-604.
Cave CB, Bryant J, Milne R. Recombinant growth hormone in children and adolescents with Turner syndrome. Cochrane Database Syst Rev.
2003(3):CD003887.
PubMed abstract
Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers. Endocr Rev.
2006;27(5):468-84.
PubMed abstract
Davenport ML. Approach to the patient with Turner syndrome. J Clin Endocrinol Metab.
2010;95(4):1487-95.
PubMed abstract
Donaldson MD, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner syndrome: from infancy to adult transfer. Arch Dis Child.
2006;91(6):513-20.
PubMed abstract / Full Text
Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner's syndrome in adulthood. Endocr Rev.
2002;23(1):120-40.
PubMed abstract
Garden AS, Diver MJ, Fraser WD. Undiagnosed morbidity in adult women with Turner's syndrome. Clin Endocrinol (Oxf).
1996;45(5):589-93.
PubMed abstract
Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, Lin AE, Mauras N, Quigley CA, Rubin K, Sandberg DE,
Sas TCJ, Silberbach M, Söderström-Anttila V, Stochholm K, van Alfen-van derVelden JA, Woelfle J, Backeljauw PF. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International
Turner Syndrome Meeting. Eur J Endocrinol.
2017;177(3):G1-G70.
PubMed abstract Endorsed by the American Academy of Pediatrics in September 2017.
Gravholt CH, Juul S, Naeraa RW, Hansen J. Prenatal and postnatal prevalence of Turner's syndrome: a registry study. BMJ.
1996;312(7022):16-21.
PubMed abstract / Full Text
Gravholt CH, Landin-Wilhelmsen K, Stochholm K, Hjerrild BE, Ledet T, Djurhuus CB, Sylvén L, Baandrup U, Kristensen BØ, Christiansen
JS. Clinical and epidemiological description of aortic dissection in Turner's syndrome. Cardiol Young.
2006;16(5):430-6.
PubMed abstract
Huang B, Thangavelu M, Bhatt S, J Sandlin C, Wang S. Prenatal diagnosis of 45,X and 45,X mosaicism: the need for thorough cytogenetic and clinical evaluations. Prenat Diagn.
2002;22(2):105-10.
PubMed abstract
Iyer NP, Tucker DF, Roberts SH, Moselhi M, Morgan M, Matthes JW. Outcome of fetuses with Turner syndrome: a 10-year congenital anomaly register based study. J Matern Fetal Neonatal Med.
2012;25(1):68-73.
PubMed abstract
Klein KO, Rosenfield RL, Santen RJ, Gawlik AM, Backeljauw PF, Gravholt CH, Sas TCJ, Mauras N. Estrogen Replacement in Turner Syndrome: Literature Review and Practical Considerations. J Clin Endocrinol Metab.
2018;103(5):1790-1803.
PubMed abstract
Lee MC, Conway GS. Turner's syndrome: challenges of late diagnosis. Lancet Diabetes Endocrinol.
2014;2(4):333-8.
PubMed abstract
Levitsky LL, Luria AH, Hayes FJ, Lin AE. Turner syndrome: update on biology and management across the life span. Curr Opin Endocrinol Diabetes Obes.
2015;22(1):65-72.
PubMed abstract
Lin AE, Basson CT, Goldmuntz E, Magoulas PL, McDermott DA, McDonald-McGinn DM, McPherson E, Morris CA, Noonan J, Nowak C,
Pierpont ME, Pyeritz RE, Rope AF, Zackai E, Pober BR. Adults with genetic syndromes and cardiovascular abnormalities: clinical history and management. Genet Med.
2008;10(7):469-94.
PubMed abstract / Full Text
Livadas S, Xekouki P, Fouka F, Kanaka-Gantenbein C, Kaloumenou I, Mavrou A, Constantinidou N, Dacou-Voutetakis C. Prevalence of thyroid dysfunction in Turner's syndrome: a long-term follow-up study and brief literature review. Thyroid.
2005;15(9):1061-6.
PubMed abstract
Loscalzo ML. Turner syndrome. Pediatr Rev.
2008;29(7):219-27.
PubMed abstract This review focuses on the diagnosis of Turner Syndrome as well as associated complications.
Mazzanti L, Cicognani A, Baldazzi L, Bergamaschi R, Scarano E, Strocchi S, Nicoletti A, Mencarelli F, Pittalis M, Forabosco
A, Cacciari E. Gonadoblastoma in Turner syndrome and Y-chromosome-derived material. Am J Med Genet A.
2005;135(2):150-4.
PubMed abstract
Menke LA, Sas TC, de Muinck Keizer-Schrama SM, Zandwijken GR, de Ridder MA, Odink RJ, Jansen M, Delemarre-van de Waal HA,
Stokvis-Brantsma WH, Waelkens JJ, Westerlaken C, Reeser HM, van Trotsenburg AS, Gevers EF, van Buuren S, Dejonckere PH, Hokken-Koelega
AC, Otten BJ, Wit JM. Efficacy and safety of oxandrolone in growth hormone-treated girls with turner syndrome. J Clin Endocrinol Metab.
2010;95(3):1151-60.
PubMed abstract
Milbrandt T, Thomas E. Turner syndrome. Pediatr Rev.
2013;34(9):420-1.
PubMed abstract
Mortensen KH, Andersen NH, Gravholt CH. Cardiovascular phenotype in Turner syndrome--integrating cardiology, genetics, and endocrinology. Endocr Rev.
2012;33(5):677-714.
PubMed abstract
Ostberg JE, Conway GS. Adulthood in women with Turner syndrome. Horm Res.
2003;59(5):211-21.
PubMed abstract
Palmer CG, Reichmann A. Chromosomal and clinical findings in 110 females with Turner syndrome. Hum Genet.
1976;35(1):35-49.
PubMed abstract
Perry RJ, Gault EJ, Paterson WF, Dunger DB, Donaldson MD. Effect of oxandrolone and timing of oral ethinylestradiol initiation on pubertal progression, height velocity and bone maturation
in the UK Turner study. Horm Res Paediatr.
2014;81(5):298-308.
PubMed abstract
Płytycz B, Espelid S, Gauperaa T, Eskeland T, Seljelid R. In vitro interactions of murine peritoneal macrophages and sarcoma cells. II. Induction of DNA synthesis in macrophages. Virchows Arch B Cell Pathol Incl Mol Pathol.
1986;50(3):293-7.
PubMed abstract
Ranke MB. Why Treat girls with Turner Syndrome with Growth Hormone? Growth and Beyond. Pediatr Endocrinol Rev.
2015;12(4):356-65.
PubMed abstract
Shankar RK, Backeljauw PF. Current best practice in the management of Turner syndrome. Ther Adv Endocrinol Metab.
2018;9(1):33-40.
PubMed abstract / Full Text
Sybert VP, McCauley E. Turner's syndrome. N Engl J Med.
2004;351(12):1227-38.
PubMed abstract / Full Text
Wiktor AE, Van Dyke DL. Detection of low level sex chromosome mosaicism in Ullrich-Turner syndrome patients. Am J Med Genet A.
2005;138A(3):259-61.
PubMed abstract
Yu B, Lu BY, Zhang B, Zhang XQ, Chen YP, Zhou Q, Jiang J, Wang HY. Overall evaluation of the clinical value of prenatal screening for fetal-free DNA in maternal blood. Medicine (Baltimore).
2017;96(27):e7114.
PubMed abstract / Full Text

 Get Help in Rhode Island
Get Help in Rhode Island

 80 KB)Click image
for full-size pdf.
80 KB)Click image
for full-size pdf.